
What is DMD?
DMD Is a Rare, X-linked, Fatal, Degenerative Neuromuscular Disorder1,2
Caused by mutations in the dystrophin gene (DMD) located on the X chromosome2
Occurs in approximately 1 in every 3500 to 5000 males born worldwide3,4
More than 90% will be nonambulant by their 2nd decade of life5
DMD is a dystrophinopathy caused by genetic changes or mutations in the 79-exon DMD gene coding for dystrophin.1,2 These genetic changes, most commonly the deletion of one or more exons, prevent the body from producing enough or any functional dystrophin.1,2

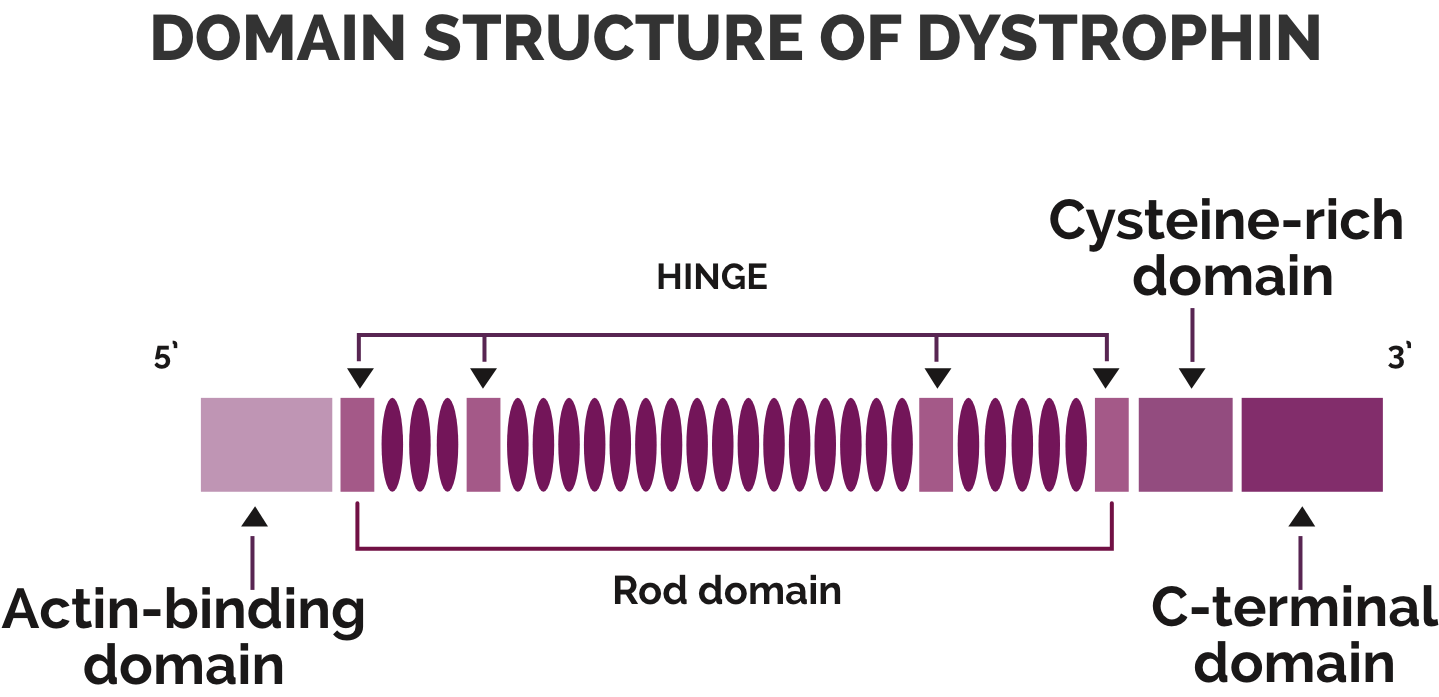
The large size (2.4 million base pairs) and structure of the DMD gene makes it susceptible to a relatively high rate of mutations, with a clustering of deletion mutations observed around the 5' region of the gene (exons 2-20) as well as the central region of the gene (exons 45-55).2,6,7 Duchenne muscular dystrophy results from DMD gene mutations leading to disruption in the normal transcript reading frame or production of a premature stop codon.6,7
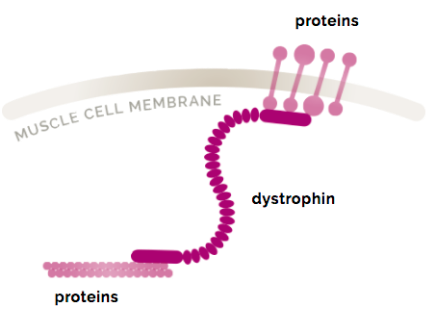
Dystrophin is one of a group of proteins that work together to strengthen muscle fibers and protect them from injury as muscles contract and relax.8 Although dystrophin exists in small amounts in the body (0.002% of total striated muscle protein),9 it plays a key structural role. Without it, muscle cells become damaged and are eventually replaced with fat and collagen.2

Disease Progression
Duchenne muscular dystrophy weakens the body’s muscles over time. And once muscle tissue is lost, it cannot be "fixed," which is why Duchenne is considered irreversible. However, some management options for Duchenne can help to slow disease progression. In the early stages of Duchenne, the disease primarily affects the muscles of the hips and thighs. This can lead to difficulty standing, climbing stairs, and maintaining balance.10
Note that the graphics show typical clinical and biological signs of DMD at various approximate age ranges. Also, DMD is a heterogeneous disease, and each child may progress differently.
0 to 4 Years
- Inflammation soon after birth11,12
- Muscle fibrosis seen as early as 1 year old11,12
- Motor delays1
- Other delays (eg, speech)1,13
5 to 7 Years
- Progressive muscle weakness14
- Enlarged calves1
- Toe walking1
- Standing-from-supine difficulty1,15
- Fat accumulation in muscle16
8 to 11 Years
- Motor milestone delays1,17
- Decreased walking ability1,17
- Part-time wheelchair use18
12 to 19 Years
- Decreased upper limb, respiratory, and cardiac function19
- Loss of ambulation18
- Ventilatory support often required17
- Unable to perform activities of daily living (ADL)20
Teens and beyond
- Increasing cardiac dysfunction19
- Heart failure17
- Life expectancy severely reduced19
Common signs of DMD can be divided into motor and
non-motor signs:
Motor signs
Not walking until
~18 months old1,21
Walking on toes with legs apart, walking with belly pointed out, or both1,14
Falling down often2
Needing help getting up from floor, or using arms to walk body to standing position (Gower’s maneuver)1,14,19
Enlarged calves19
Fatigue22
Non-motor signs
Emotional/behavioral challenges23
Aggressive/noncompliant behavior, depressed/overcontrolled behavior
Cognitive impairment13,23
Intellectual disability, learning disability, delayed speech
Neuropsychiatric disorders23
ADHD, anxiety, autism spectrum disorder, epilepsy